Google's DeepMind AI Predicts 3D Structure of Nearly Every Protein Known to Science
At last, the decades-old protein folding problem may finally be put to rest.


This ribbon diagram shows the 3D protein structure of an antibody. Complex? It's pretty simple for an AI.
It wasn't until 1957 when scientists earned special access to the molecular third dimension.
After 22 years of grueling experimentation, John Kendrew of Cambridge University finally uncovered the 3D structure of a protein. It was a twisted blueprint of myoglobin, the stringy chain of 154 amino acids that helps infuse our muscles with oxygen. As revolutionary as this discovery was, Kendrew didn't quite open up the protein architecture floodgates. During the next decade, fewer than a dozen more would be identified.
Fast-forward to today, 65 years since that Nobel Prize-winning breakthrough.
On Thursday, Google's sister company, DeepMind, announced it has successfully used artificial intelligence to predict the 3D structures of nearly every catalogued protein known to science. That's over 200 million proteins found in plants, bacteria, animals, humans — almost anything you can imagine.
"Essentially, you can think of it as covering the entire protein universe," Demis Hassabis, founder and CEO of DeepMind, told reporters this week.
It's thanks to AlphaFold, DeepMind's groundbreaking AI system, which has an open-source database so scientists worldwide can involve it in their research at will, and for free. Since AlphaFold's official launch in July of last year — when it had only pinpointed some 350,000 3D proteins — the program has made a noticeable dent in the landscape of research.
"More than 500,000 researchers and biologists have used the database to view over 2 million structures," Hassabis said. "And these predictive structures have helped scientists make brilliant new discoveries."
In April, for instance, Yale University scientists called on AlphaFold's database to aid in their goal of developing a new, highly effective Malaria vaccine. And in July of last year, University of Portsmouth scientists used the system to engineer enzymes that will fight against single-use plastic pollution.
"This moved us a year ahead of where we were, if not two," John McGeehan, director of Portsmouth's Center for Enzyme Innovation and a researcher behind the latter study, told the New York Times.
The 3D structure of vitellogenin, which makes up egg yolk.
These endeavors are just a small sample of AlphaFold's ultimate reach.
"In the past year alone, there have been over a thousand scientific articles on a broad range of research topics which use AlphaFold structures; I have never seen anything like it," Sameer Velankar, DeepMind collaborator and team leader at the European Molecular Biology Laboratory's Protein Data Bank, said in a press release.
Others who've used the database, according to Hassabis, include those trying to improve our understanding of Parkinson's disease, people hoping to protect the health of honeybees and even some looking to gain valuable insight into human evolution.
"AlphaFold is already changing the way we think about the survival of molecules in the fossil record, and I can see it will soon become a fundamental tool for researchers working not only in evolutionary biology but also in archaeology and other palaeo-sciences," Beatrice Demarchi, an associate professor at the University of Turin, who recently used the system in a study on an ancient egg controversy, said in a press release.
In the coming years, DeepMind also intends to partner with teams at the Drugs For Neglected Diseases Initiative and the World Health Organization, with the goal of finding cures for little-studied, yet pervasive, tropical diseases such as Chagas disease and Leishmaniasis.
"It will make many researchers around the world think about what experiments they could do," Ewan Birney, DeepMind collaborator and deputy director of the EMBL, told reporters. "And think about what is going on in the organisms and the systems that they study."
Locks and keys
So, why do so many scientific advancements depend on this treasure chest of 3D protein modeling? Let's explain.
Suppose you're trying to make a key that fits perfectly into a lock. But you have no way of viewing the structure of that lock. All you know is this lock exists, some data about its materials, and maybe numerical information on how big each ridge is and sort of where those ridges ought to be.
Developing this key wouldn't be impossible, maybe, but it'd be quite difficult. Keys have to be precise, otherwise they don't work. Therefore, before you get started, you'd probably try your best to model a few different mock locks with whatever info you do have so you can make your key.
In this analogy, the lock is a protein and the key is a small molecule that binds to this protein.
For scientists, whether they're doctors trying to craft novel medications or botanists dissecting plant anatomy to make fertilizers, interplay between certain molecules and proteins is crucial.
With medications, for instance, the specific way a molecule in a drug binds to a protein could be the breaking point for whether it works. This interaction gets complicated because even though proteins are just strings of amino acids, they're not straight or flat. They inevitably fold, bend and sometimes tangle around themselves, like headphone wires in your pocket.
In fact, a protein's unique folds dictate how it functions — and even the slightest of folding mistakes in the human body can lead to disease.
But returning to small molecule medications, sometimes pieces of a folded protein are blocked from binding a drug. They might happen to be folded in a strange way that makes them inaccessible, for instance. Things like this are very important bits of information for scientists trying to get their drug molecule to stick. "I think it's true that almost every drug that has come to market over the past few years has been, in part, designed through knowledge of protein structures," Janet Thornton, a research scientist at the EMBL, said in the conference.
This is why researchers normally spend an incredible amount of time and effort to decode the folded, 3D structure of a protein they're working with in the way you'd begin your key-making journey by piecing together the lock's mould. If you know the exact structure, it becomes a lot easier to tell where and how a molecule would attach to a given protein, as well as how that attachment might affect the protein's folds in response.
But this endeavor isn't simple. Or cheap.
"The cost of solving a new, unique structure is on the order of $100,000," Steve Darnell, a structural and computational biologist from the University of Wisconsin and researcher at bioinformatics company DNAStar, said in a statement.
That's because the solution typically comes from super complicated laboratory experiments.
Kendrew, for example, tapped into a technique called X-ray crystallography back in the day. Basically, this method requires you to take solid crystals of the protein you're interested in, place them in an X-ray beam, and watch to see what pattern the beam makes. That pattern is pretty much the position of thousands of atoms within the crystal. Only then can you use the pattern to uncover a protein's structure.
There's also the more recent technique known as cryo-electron microscopy. This one's similar to X-ray crystallography, except the protein sample gets straight-up blasted with electrons instead of an X-ray beam. And even though it's considered much higher in resolution than the other technique, it can't exactly penetrate everything. Further, in the realm of technology, some have attempted to digitally create protein folding structures. But early tries, like a few attempts in the '80s and '90s were not great. As you can imagine, laboratory methods are also tedious — and difficult.
Over the years, such barriers have given rise to what's called the "protein folding problem." Simply, scientists don't know how proteins fold, and have faced significant hurdles to get past that issue.
AlphaFold's AI could be a game changer.
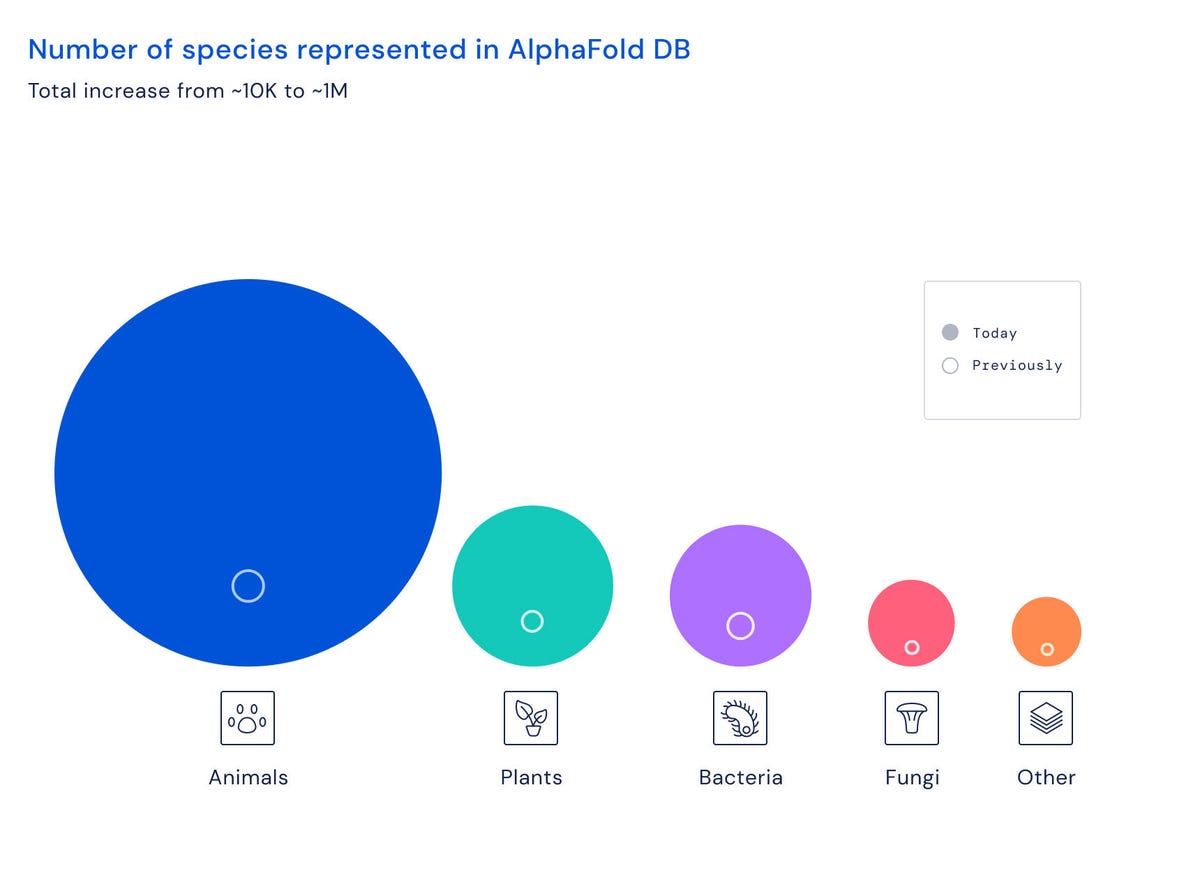
A diagram provided by DeepMind of the explosive growth of the AlphaFold database, by species.
Solving the 'folding problem'
In short, AlphaFold was trained by DeepMind engineers to predict protein structures without requiring laboratory presence. No crystals, no electron firing, no $100,000 experiments.
To get AlphaFold to where it is today, first, according to the company's website, the system was exposed to 100,000 known protein folding structures. Then, as time passed, it started to learn how to decode the rest.
It's really as straightforward as that. (Well, apart from the talent that went into coding the AI.)
"It takes, I don't know, a minimum of $20,000 and a large amount of time to crystallize a protein," Birney said. "That means experimentalists have to make choices about what they do – AlphaFold hasn't had to make choices yet." This feature of AlphaFold's thoroughness is quite fascinating. What this means is scientists have more liberty to guess and check, follow an inkling or gut instinct and cast a wide net in their research when it comes to protein structures. They won't need to worry about cost or timelines.
"The models come with a prediction error as well," Jan Kosinski, DeepMind collaborator and structural modeler at the EMBL in Hamburg, Germany said. "And usually — actually in many cases — the error is really tiny. So we call that a near-atomic precision."
Further, the DeepMind team also says it conducted a wide variety of risk assessments to make sure using AlphaFold is safe and ethical. DeepMind team members also suggested that AI, in general, might carry biosecurity risks we hadn't thought to assess before — especially as such technology continues to permeate the medical space.
But as the future unfolds, the DeepMind crew says AlphaFold will fluidly adapt and address such worries on a case-by-case basis. For now, it seems to be working — with a universe of protein models tracing back to a modest portrait of myoglobin.
"Only two years ago," Birney said, "we just simply did not realize that this was feasible."
Correction at 6:45 a.m. PT: Janet Thornton's last name and title have been fixed.